Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
МКБ-10
Общие сведения
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Причины
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Классификация
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
Симптомы
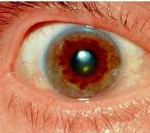
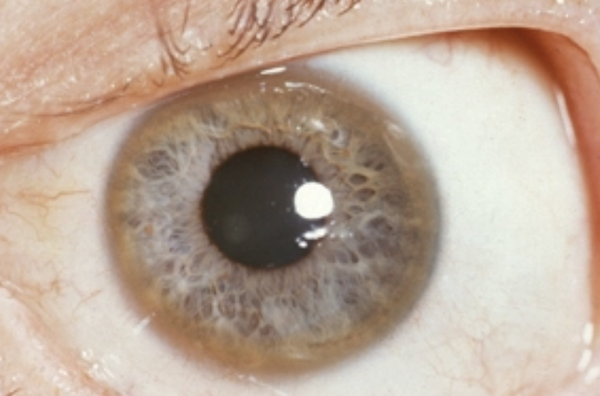
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Диагностика
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди. Для подтверждения болезни Вильсона проводится генодиагностика.
Лечение болезни Вильсона
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Нарушения обмена меди (болезнь Вильсона) у детей
Общая информация
Краткое описание
Союз педиатров России
Год утверждения (частота пересмотра): 2016 (пересмотр каждые 3 года)

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Е 83.0 – Нарушения обмена меди.
• Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжелым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально 3.
Этиология и патогенез
ATP7B – ген экспрессируется, в основном, в печени и кодирует медьтранспортирующую АТФ-азу (АТФ-аза 7В, АТФ-аза 2, АТФ-аза Р-типа). Генетически детерминируемое снижение функции медь-транспортирующей АТФ-азы в результате молекулярных дефектов в гене АТР7В приводит к снижению гепатобилиарной экскреции меди и нарушению встраивания меди в церулоплазмин, в результате экскретируется и циркулирует апоцерулоплазмин (ненагруженный медью, срок полувыведения которого сокращается вдвое, что и объясняет гипоцерулоплазминемию), а медь накапливается в различных органах и тканях, преимущественно в печени, головном мозге, роговице глаза, почках, обеспечивая полиморфизм клинических появлений БВ. Вся циркулирующая (в сыворотке крови) медь связана церулоплазмином, а парадокс БВ, при которой отмечается низкий уровень меди в сыворотке крови при перегрузке тканей, объясняется низким уровнем церулоплазмина. При тяжелых формах БВ, протекающих с цитолизом, повышение концентрации меди до нормы и выше связано с распадом перегруженных медью гепатоцитов, свободная (не связанная церулоплазмином) медь крайне токсична и провоцирует гемолитические кризы 5.
| Продукт | Медь, мг в 100г | Продукт | Медь, мг в 100г | |
| Печень телячья жареная | 23,9 | Малина | 0,170 | |
| Печень баранья жареная | 13,5 | Абрикос | 0,170 | |
| Устрицы | 7,5 | Редис | 0,150 | |
| Угри вареные | 6,6 | Яйца куриные | 0,150 | |
| Дрожжи сухие | 5,0 | Картофель | 0,140 | |
| Какао-порошок | 3,9 | Свекла | 0,140 | |
| Пюре томатное | 2,9 | Баклажан | 0,135 | |
| Семена подсолнечника | 2,3 | Киви | 0,135 | |
| Орехи кешью | 2,1 | Чеснок | 0,130 | |
| Креветки вареные | 1,9 | Крыжовник | 0,130 | |
| Крабы вареные | 1,8 | Смородина черная | 0,130 | |
| Орехи бразильские | 1,8 | Земляника садовая | 0,125 | |
| Семена тыквенные | 1,6 | Салат | 0,120 | |
| Семена кунжута | 1,5 | Груша | 0,120 | |
| Тахини | 1,5 | Капуста брокколи | 0,120 | |
| Омары вареные | 1,4 | Яблоки | 0,110 | |
| Орехи грецкие | 1,3 | Помидор | 0,110 | |
| Орехи кедровые | 1,3 | Редька | 0,100 | |
| Фундук | 1,2 | Огурцы | 0,100 | |
| Арахис | 1,0 | 0,8 | Слива | 0,087 |
| Смородина | 0,8 | Лук репчатый | 0,085 | |
| Горошек зеленый | 0,75 | Морковь | 0,080 | |
| Арахисовое масло | 0,7 | Виноград | 0,080 | |
| Грибы | 0,7 | Капуста зеленая | 0,075 | |
| Чечевица | 0,66 | Сыр Чеддер | 0,070 | |
| Греча ядрица | 0,64 | Сыр российский | 0,070 | |
| рис | 0,560 | Сыр рассольный | 0,070 | |
| геркулес | 0,450 | Апельсин | 0,067 | |
| кукуруза | 0,290 | Сыр адыгейский | 0,060 | |
| лимон | 0,240 | Дыня | 0,047 |
Невролог «СМ-Клиника» рассказала о течении болезни Вильсона-Коновалова у взрослых
В 1912 году одновременно в нашей стране и за рубежом была описана особая наследственная патология, которая получила свое название по авторам — болезнь Вильсона-Коновалова. Это наследственная болезнь и она опасна. Можно ли ее вылечить — выясним с экспертом.

АЛЕНА ПАРЕЦКАЯ
Врач-патофизиолог, иммунолог, член
Санкт-Петербургского общества патофизиологов
ВАЛЕНТИНА КУЗЬМИНА
К.м.н., врач-невролог «СМ-Клиника»
Один из самых характерных признаков болезни – это патологическое накопление меди в области различных органов, повреждение тканей, особенно – печени, проблемы нервной системы, изменения в радужке глаза.
Что нужно знать о болезни Вильсона-Коновалова
Что такое болезнь Вильсона-Коновалова
Термином болезнь Вильсона-Коновалова называют наследственную патологию. Она возникает, когда родители передают ребенку дефектный ген (АТР7В). Состояние относится к аутосомно-рецессивным патологиям, то есть возникает, если каждый из родителей несет подобный ген в своих клетках и ребенок наследует сразу оба гена – от матери и от отца.
Этот дефектный ген дает команды к синтезу белка, который регулирует обмен и перенос меди внутри организма. При его дефекте медь копится в печени, концентрируется в нервных ганглиях, откладывается в радужке глаза. Патология встречается нечасто, ее иногда очень сложно распознать, особенно, если в семье нет подобных больных.
Причины болезни Вильсона-Коновалова у взрослых
Ключевой процесс при этой патологии – наследование дефектного гена от родителей. Он располагается в 13-ой хромосоме и регулирует обмен меди.
Подавляющий объем микроэлемента (95%) переносится в тесной связке с белком плазмы – церулоплазмином. Его постоянно образует печень, и только около 5% меди переносится вместе с альбумином.
Медь нужна для участия в метаболических процессах, в том числе – окислительных. Если развивается болезнь Вильсона, выведение ее нарушается, повышается концентрация в плазме, оттуда она разносится в ткани. Основное накопление меди происходит в мозге, в области радужки, внутри печени, а также в почках. Избыток микроэлемента оказывает токсический эффект.
Симптомы болезни Вильсона-Коновалова у взрослых
При брюшной форме болезни симптомы обычно проявляются ближе к 40 годам. Среди ключевых признаков:
Самая редкая форма болезни – это экстрапирамидно-корковые расстройства. Он похожи на все формы, дополнительно будут судорожные приступы, выраженные проблемы интеллекта, расстройства движений.
Лечение болезни Вильсона-Коновалова у взрослых
Диагностика
Если речь идет о проявлении глазных симптомов, врач предварительно осматривает состояние глаз щелевой лампой для того, чтобы подтвердить наличие кольца Кайзера-Флейшера.
Показано назначение биохимических тестов крови и мочи, которые покажут повышенное содержание меди в моче и сниженную концентрацию церулоплазмина в плазме крови.
КТ или МРТ покажут атрофические процессы в области мозга и мозжечка, повреждение базальных ядер.
Дополнительно проводится консультация генетика и ряд генетических тестов, выявляющих дефектные гены.
Современные методы лечения
Основной метод лечения при этой болезни – назначение тиоловых препаратов, особенно – унитиола либо D-пеницилламина, купренила. Препараты принимают длительно, врач подбирает наиболее оптимальную дозу, которая позволит избежать побочных эффектов.
Дополнительно врач может применять препараты из группы нейролептиков, при ригидности мышц – леводопу или карбидопу.
При тяжелом течении показана трансплантация печени, иммунсупрессивная терапия. Возможно применение биогемоперфузии с изолятом живых клеточных элементов селезенки с печенью.
Дополнительно необходимо соблюдение диеты с исключением продуктов, содержащих большое количество меди.
Профилактика болезни Вильсона-Коновалова у взрослых в домашних условиях
– Для профилактики патологии, – говорит врач-невролог Валентина Кузьмина, – необходимо придерживаться диеты №5, а также ограничить потребление меди до 1 г в сутки – исключить орехи, сухофрукты, шоколад, раков, печенье, цельную пшеницу. Также рекомендован прием препаратов группы витамина В6, унитиола, триентина.
Популярные вопросы и ответы
Мы поговорили о проблемах болезни Вильсона-Коновалова, ее осложнениях и возможности самолечения с врачом-неврологом Валентиной Кузьминой.
Какие могут последствия при болезни Вильсона-Коновалова?
Среди основных последствий болезни Вильсона-Коновалова можно выделить:
Вызвать врача на дом необходимо при нарушении речи (дизартрия) и глотания (дисфагия), насильственном непроизвольном смехе или плаче, нарушении эмоционального состояния, умеренном снижении интеллекта.
Можно ли вылечить болезнь Вильсона-Коновалова народными средствами?
Нет, лечить болезнь Вильсона-Коновалова народными средствами ни в коем случае нельзя. Это только навредит и ухудшит проблемы печени и нервной системы. Обязательно обратитесь к специалисту.
Болезнь Вильсона у детей. Клинические рекомендации.
Болезнь Вильсона у детей
Оглавление
Ключевые слова
Список сокращений
БВ – болезнь Вильсона
МНО – международное нормализованное отношение
ОТП – ортотопическая трансплантация печени
ПИ – протромбиновый индекс
УЗИ – ультразвуковое исследование
ЩФ – щелочная фосфатаза
МРТ – магнитно-регонансная томография
КТ – компьютерная томография
Термины и определения
Хелатная терапия – способ лечения, заключающийся во введении препаратов, связывающих и выводящих из организма ионы тяжелых металлов.
Гепатомегалия – увеличение печени.
Спленомегалия – увеличение селезенки.
Стеатоз печени – наиболее распространенный гепатоз, при котором в печёночных клетках происходит накопление жира.
Фиброз печени – разрастание соединительной ткани органа, возникающее при чрезмерном накоплении белков внеклеточного матрикса (основы соединительной ткани).
Цирроз печени – хроническое заболевание печени, сопровождающееся необратимым замещением паренхиматозной ткани печени фиброзной соединительной тканью, или стромой.
1. Краткая информация
1.1 Определение
Болезнь Вильсона (БВ) относится к числу наиболее трудно диагностируемых заболеваний печени в связи с длительным латентным течением, особенно на начальных стадиях заболевания, и большим полиморфизмом клинической симптоматики. В связи с этим болезнь Вильсона необходимо исключать у каждого пациента детского и подросткового возраста с патологией печени неуточненной этиологии.
Своевременное назначение патогенетической терапии при болезни Вильсона у детей сопровождается регрессом клинической симптоматики, предотвращением формирования цирроза печени и неврологической симптоматики, улучшением качества жизни и социальной адаптации ребенка. В связи с этим важнейшей медицинской и социальной задачей является ранняя диагностика и адекватная терапия БВ 4.
Болезнь Вильсона (синонимы: болезнь Вильсона-Коновалова, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – редкое наследственное заболевание, связанное с нарушением метаболизма меди и избыточным ее накоплением в различных органах и тканях, преимущественно проявляющееся симптоматикой поражения печени и центральной нервной системы 2.
1.2 Этиология и патогенез
ATP7B – ген экспрессируется, в основном, в печени и кодирует медьтранспортирующую АТФ-азу (АТФ-аза 7В, АТФ-аза 2, АТФ-аза Р-типа). Генетически детерминируемое снижение функции медь-транспортирующей АТФ-азы в результате молекулярных дефектов в гене АТР7В приводит к снижению гепатобилиарной экскреции меди и нарушению встраивания меди в церулоплазмин, в результате экскретируется и циркулирует апоцерулоплазмин (ненагруженный медью, срок полувыведения которого сокращается вдвое, что и объясняет гипоцерулоплазминемию), а медь накапливается в различных органах и тканях, преимущественно в печени, головном мозге, роговице глаза, почках, обеспечивая полиморфизм клинических появлений БВ. Вся циркулирующая (в сыворотке крови) медь связана церулоплазмином, а парадокс БВ, при которой отмечается низкий уровень меди в сыворотке крови при перегрузке тканей, объясняется низким уровнем церулоплазмина. При тяжелых формах БВ, протекающих с цитолизом, повышение концентрации меди до нормы и выше связано с распадом перегруженных медью гепатоцитов, свободная (не связанная церулоплазмином) медь крайне токсична и провоцирует гемолитические кризы 10.
В организм медь поступает в основном с пищей (табл.1). Наиболее богаты медью следующие продукты: печень, моллюски, крабы, креветки, устрицы, лобстеры, соевые бобы, шоколад, орехи.
Болезнь Вильсона-Коновалова у детей
Общая информация
Краткое описание
Одобрен объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения Республики Казахстан
от «28» ноября 2017 года
Протокол №33
Болезнь Вильсона-Коновалова (синонимы гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – тяжелое прогрессирующее наследственное заболевание, передающееся по аутосомно-рецессивному типу, в основе которого лежит нарушение экскреции меди из организма, приводящее к избыточному накоплению этого микроэлемента в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (преимущественно подкорковых ядер).
NB! Причиной возникновения БВК являются мутации гена ATP7B, который локализован на 13 хромосоме в локусе 13q14.3 и кодирует медь транспортирующую АТФ-азу Р-типа – ATP7B.
Код(ы) МКБ-10:
| МКБ-10 | |
| Код | Название |
| E83.0 | Нарушение обмена меди (Болезнь Вильсона-Коновалова) |
Дата разработки/пересмотра протокола: 2017 год.
Сокращения, используемые в протоколе:
| анти-LC | антитела к цитозольному антигену печени |
| анти-LKM | антитела к микросомам печени и почек |
| анти-LP | антитела к белкам печени и поджелудочной железы |
| анти-SLA | антитела к растворимым печеночным антигенам |
| БВК | болезнь Вильсона-Коновалова |
| БХАК | биохимический анализ крови |
| ГГТП | Гамма-глютамилтранспептидаза |
| ГЛД | гепатолентикулярная дегенерация |
| КТ | компьютерная томография |
| МРТ | магниторезонансная томография |
| ОАК | общий анализ крови |
| ОАМ | общий анализ мочи |
| СОЭ | скорость оседания эритроцитов |
| СРБ | С-реактивный протеин |
| УЗИ | ультразвуковое исследование |
| ЩФ | Щелочная фосфотаза |
| ЭКГ | электрокардиограмма |
| ЭНМГ | электронейромиография |
| ЭРХПГ | Эндоскопическая ретроградная холангиопанкреатография |
| ЭЭГ | электроэнцефалография |
| ANA | антинуклеарные антитела |
| IgG | иммуноглобулин G |
| АNCА | антитела к цитоплазме нейтрофилов |
| АМА | антимитохондриальные антитела |
Пользователи протокола: врачи общей практики, педиатры, детские гастроэнтерологи, детские неврологи.
Категория пациентов: дети.
Шкала уровня доказательности:
| A | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| B | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| C | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |
Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран
Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Клиническая картина гепатолентикулярной дегенерации характеризуется большим полиморфизмом в отношении как неврологических, так и соматических проявлений. Этот полиморфизм отражен в различных классификациях заболевания.
Формы болезни Вильсона [3]:
· Бессимптомная форма;
· Печеночная форма;
· Церебральная форма;
· Смешанная форма.
В зависимости от вовлечения в патологический процесс печени и центральной нервной системы и характера экстрапирамидной симптоматики, распознают 5 форм гепато-церебральной дистрофии [11]:
· Брюшная (абдоминальная) форма – манифестирует в возрасте от 5 до 17 лет и характеризуется различными вариантами поражения печени, нередко принимающими злокачественное «галопирующее» течение, приводящее к смерти раньше появления симптомов со стороны нервной системы. Её продолжительность от нескольких месяцев до 3-5 лет.
· Ригидно-аритмогиперкинетическая, или ранняя форма отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
· Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возраста, протекает медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжѐлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
· Дрожательная форма начинается в возрасте 20-30 лет, протекает довольно медленно (10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжѐлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
· Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепатоцеребральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжѐлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-6,10,19,20]: на БВ должны обследоваться дети в возрасте от 2 до 18 лет, имеющих необъяснимое повышение сывороточных аминотрансфераз, проявления фульминантной печеночной недостаточности, хронического гепатита, цирроза печени, неврологические нарушения неустановленной этиологии, Кумбс-негативную гемолитическую анемию, отягощенный семейный анамнез по БВ. Диагностика БВ базируется на комбинации клинических симптомов, данных лабораторного обследования и молекулярно-генетического тестирования.
Диагностические критерии [1-4]
Жалобы:
· боли в животе различной локализации;
· изменение цвета кожи;
· носовые кровотечения;
· тремор и непроизвольные движения;
· слюнотечение, дизартрия, нарушение глотания;
· мигренеподобные головные боли;
· бессонница;
· депрессия;
· невротическое поведение;
· изменения личности;
· психоз.
Анамнез: Первичная манифестация БВ может протекать в виде острого фульминантного гепатита, проявляющегося коагулопатией, энцефалопатией, Кумбс-негативной гемолитической анемией, печеночноклеточной и почечной недостаточностью, с выявлением значительного превышения меди в сыворотке крови и моче.
NB! Обратить внимание на возраст начала проявлений заболевания у пациента: до 5 летнего возраста проявления болезни Вильсона-Коновалова, как правило, отсутствуют. Болезнь манифестирует в возрасте 8-16 лет (хотя практически с рождения отмечается повышенная активность печеночных аминотрансфераз). Необходимо уточнить о наличии заболеваний печени и нейропсихических нарушений у ближайших родственников больного (стеатоза, гепатитов, цирроза печени, печеночной недостаточности).
NB! Первые симптомы заболевания начинается с симптомов поражения печени (в 42% случаев). Примерно у 25% пациентов заболевание начинается остро, с развития желтухи, астенического синдрома, анорексии, повышения температуры. Болезнь Вильсона-Коновалова может клинически протекать по типу аутоиммунного гепатита, с повышением уровня сывороточных иммуноглобулинов и неспецифических аутоантител, в связи с чем необходимо исключать данное заболевание и у больных с аутоиммунным гепатитом. Неврологическая и психическая симптоматика наблюдается у 10% больных. У 15% пациентов болезнь Вильсона-Коновалова манифестирует гематологическими синдромами (прежде всего гемолитической анемией). Кольцо Кайзера-Флейшера не выявляется у детей до 5 лет.
| Проявления болезни Вильсона-Коновалова | Симптомы |
| Поражение печени | Бессимптомная гепатомегалия |
| Изолированная спленомегалия | |
| Стеатогепатит | |
| Острый (фульминантный) гепатит | |
| Аутоиммуноподобный гепатит | |
| Цирроз печени | |
| Поражение ЦНС | Двигательные нарушения (тремор, непроизвольные движения) |
| Слюнотечение, дизартрия | |
| Ригидная дистония | |
| Псевдобульбарный синдром | |
| Вегетососудистая дистония | |
| Мигренеподобные головные боли | |
| Бессоница | |
| Дистонические атаки | |
| Психиатрические симптомы | Депрессия |
| Невротическое поведение | |
| Изменения личности | |
| Психоз | |
| Другие системы | Офтальмология: кольца Кайзера-Флейшера, «медная» катаракта |
| Гемолитическая анемия | |
| Патология почек: аминоацидурия, нефролитиаз | |
| Патология скелета: ранний остеопороз, артрит | |
| Поражение сердца: кардиомиопатия, нарушения ритма | |
| Панкреатит, желчекаменная болезнь | |
| Гипопаратиреодизм, гигантизм |
Физикальное обследование 3:
необходимо оценить наличие:
· смуглого («медный») цвета кожи;
· желтушности склер;
· незначительной или умеренной гепатомегалии;
· спленомегалии;
· неврологических нарушений и психических расстройств в виде непроизвольных движений в мышцах торса и конечностей;
· мигренеподобной головной боли;
· скованности в мышцах;
· эмоциональной лабильности;
· агрессивности.
Лабораторные исследования 4:
· общий анализ крови: лейкопения, нормохромная анемия, тромбоцитопения, ретикулоцитоз, ускоренная СОЭ.
· общий анализ мочи: при поражении почек можно обнаружить микрогематурию, незначительную протеинурию, гиперкальциурию.
· суточная экскреция мочи: гиперкупренилурия, признаки развившейся тубулопатии с признаками: глюкозурией, аминоацидурией, фосфатурией, уратурией, протеинурией.
· биохимический анализ крови: снижение церулоплазмина и общей меди, увеличение уровней свободной меди (таблица 1), аминотрасфераз (в 1,5-50 раз); билирубин повышен более чем в 2 раза, преимущественно за счет прямой фракции; уровень щелочной фосфатазы обычно повышен; может быть повышена активность гаммаглютамилтранспептидазы (ГГТП); гипоальбуминемия.
· коагулограмма: снижение протромбинового индекса, гипофибриногенемия, снижение тромбинового времени.
· пеницилламиновый тест: необходимо исследовать мочу, собранную сразу после приема 500 мг пеницилламина и через 12 часов. У пациентов с болезнью Вильсона-Коновалова суточная экскреция меди будет повышаться до более 1500 мкг/дл/сут (норма
| Показатель | Норма | ГЛД |
| Церулоплазмин, мг/дл | 17–40 | |
| Общая медь в сыворотке крови, мкмоль/л | 12–32 | |
| Свободная медь в сыворотке крови, мкг/дл | 5–12 | > 50 |
| Суточная экскреция меди с мочой, мкг/сут | > 100–1000 | |
| Суточная экскреция меди с мочой в пробе с Д-пеницилламином, мкг/сут | 600–800 | 1000–3000 |
| Количественное содержание меди в печени, мкг/г | 15–55 | > 250 |
Показания для консультации специалистов 2:
· консультация офтальмолога – для выявления колец Кайзера-Флейшера, также на наличие катаракты у детей, с целью исключения других болезней накопления;
· консультация невропатолога – оценка неврологического статуса, нервно-психического статуса;
· консультация психиатра – диагностика психиатрических состояний;
· консультация психотерапевта – коррекция психологических проблем;
· консультация гастроэнтеролога – коррекция нарушений желудочно-кишечного тракта;
· консультация гематолога – при наличии симптомов гемолитической анемии, коагулопатии;
· консультация нефролога – при наличии патологии в анализах мочи;
· консультация сурдолога – определение остроты слуха;
· консультация физиотерапевта – определение методов физиотерапевтического лечения;
· консультация хирурга – при риске пищеводно-желудочных кровотечений, для выявления показаний к проведению трансплантации печени у детей с признаками цирроза печени (ЦП), печеночно-клеточной декомпенсацией;
· консультация инфекциониста – при наличии сопутствующего вирусного гепатита;
· консультация отоларинголога – при инфекциях верхних дыхательных путей.
Диагностический алгоритм: 
Рисунок 1- Алгоритм диагностики болезни Вильсона [3]. КФ – кольца Кайзера-Флейшера; Цер – церулоплазмин; 24-h Cu – суточная экскреция меди с мочой.
Ни один лабораторный тест (за исключением полного секвенирования патологического гена АТР7В) не обладает 100% чувствительностью и не обеспечивает 100% специфичность, таблица 3.
| Тест | Характерные находки | Ложноотрицательный результат | Ложноположительный результат |
| Сывороточный церулоплазмин | Уменьшение на 50% относительно нормы | Нормальный уровень у пациентов с выраженным воспалением в печени. Завышение результата при иммунологических методах исследования. Беременность, прием эстрогенов. | Низкий уровень при: -мальабсорбции; -ацерулоплазминемии; -гетерозиготы |
| Суточная экскреция меди с мочой | >0,64мкмоль/24часа | Норма: -неправильный сбор мочи; -дети без вовлечения печени | Повышение: -гепатоцеллюлярный некроз; -холестаз; -контаминация |
| Свободная медь сыворотки | >1,6мкмоль/л | Норма при завышении уровня церулоплазмина иммунологическими методами | |
| Печеночная медь | >4мкмоль/г сухого вещества | В зависимости от места взятия материала: -активная болезнь печени; -узелки регенерации | Синдромы холестаза |
| Кольцо Кайзера-Флейшнера (при использовании щелевой лампы) | Наличие кольца | Отсутствует: -у 50% пациентов с печеночной формой; -у большинства асимптоматических сибсов | Первичный биллиарный цирроз |
Диагноз болезни Вильсона ставится на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа (таблица 4).
| Типичные клинические симптомы и признаки | Другие тесты |
| Кольцо Кайзера-Флейшнера: Присутствует 2 Отсутствует 0 | Печеночная медь (при отсутствии холестаза) >5х верхней границы нормы (>4мкМ/г) 2 0,8-4мкМ/г 1 Норма ( |
| Неврологическая симптоматика или типичные изменения на МРТ головного мозга: Тяжелая 2 Средняя 1 Отсутствует 0 | Медь в моче (в отсутствии острого гепатита) Норма 0 1-2х верхней границы нормы 1 >2х верхней границы нормы 2 Норма, но увеличение >5х верхней границы нормы после пеницилламина 2 |
| Церулоплазмин сыворотки Норма (>0,2г/л) 0 0,1-0,2 г/л 1 | Анализ мутаций Обе хромосомы 4 На одной хромосоме 1 Не выявлено мутаций 0 |
| Кумбс-негативная гемолитическая анемия Присутствует 1 Отсутствует 0 | |
| Интерпретация результата | |
| 4 и более | Диагноз установлен |
| 3 | Диагноз возможен, необходимы дальнейшие исследования |
| 2 и менее | Диагноз маловероятен |
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований 15
Таблица 5– Дифференциальный диагноз болезни Вильсона-Коновалова.
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Клоназепам (Clonazepam) |
| Пантопразол (Pantoprazole) |
| Пеницилламин (Penicillamine) |
| Пиридоксин (Pyridoxine) |
| Ранитидин (Ranitidine) |
| Тиаприд (Tiapride) |
| Тригексифенидил (Trihexyphenidyl) |
| Триентин (Trientine) |
| Цинка ацетат (Zinc acetate) |
| Цинка оксид (Zinc oxide) |
| Цинка сульфат (Zinc sulfate) |
| Эзомепразол (Esomeprazole) |
Лечение (амбулатория)
ТАКТИКА ЛЕЧЕНИЯ НА АМБУЛАТОРНОМ УРОВНЕ [1-4,11]:
· поддерживающая хелатная терапия;
· диспансерное наблюдение.
Лечение (стационар)
ТАКТИКА ЛЕЧЕНИЯ НА СТАЦИОНАРНОМ УРОВНЕ [1-4,9-11]: все пациенты с данным диагнозом лечатся на стационарном уровне. Проводится подбор хелатной терапии с учетом индивидуальных особенностей пациента, подбор/коррекция хелатной терапии и подбор симптоматической терапии, м при необходимости ортотопическая трансплантация печени.
Карта наблюдения пациента, маршрутизация пациента:
| Продукт | Медь, мг в 100г | Продукт | Медь, мг в 100г |
| Печень телячья жареная | 23,9 | Малина | 0,170 |
| Печень баранья жареная | 13,5 | Абрикос | 0,170 |
| Устрицы | 7,5 | Редис | 0,150 |
| Угри вареные | 6,6 | Яйца куриные | 0,150 |
| Дрожжи сухие | 5,0 | Картофель | 0,140 |
| Какао-порошок | 3,9 | Свекла | 0,140 |
| Пюре томатное | 2,9 | Баклажан | 0,135 |
| Семена подсолнечника | 2,3 | Киви | 0,135 |
| Орехи кешью | 2,1 | Чеснок | 0,130 |
| Креветки вареные | 1,9 | Крыжовник | 0,130 |
| Крабы вареные | 1,8 | Смородиначерная | 0,130 |
| Орехи бразильские | 1,8 | Земляника садовая | 0,125 |
| Семена тыквенные | 1,6 | Салат | 0,120 |
| Семена кунжута | 1,5 | Груша | 0,120 |
| Тахини | 1,5 | Капуста брокколи | 0,120 |
| Омары вареные | 1,4 | Яблоки | 0,110 |
| Орехи грецкие | 1,3 | Помидор | 0,110 |
| Орехи кедровые | 1,3 | Редька | 0,100 |
| Фундук | 1,2 | Огурцы | 0,100 |
| Арахис | 1,0 | Перец красный сладкий | 0,100 |
| Кальмары | 1,0 | Вишня | 0,100 |
| Миндаль | 1,0 | Лук зеленый | 0,092 |
| Фисташки жареные соленые | 0,8 | Слива | 0,087 |
| Смородина | 0,8 | Лук репчатый | 0,085 |
| Горошек зеленый | 0,75 | Морковь | 0,080 |
| Арахисовое масло | 0,7 | Виноград | 0,080 |
| Грибы | 0,7 | Капуста зеленая | 0,075 |
| Чечевица | 0,66 | Сыр Чеддер | 0,070 |
| Греча ядрица | 0,64 | Сыр российский | 0,070 |
| рис | 0,560 | Сыр рассольный | 0,070 |
| геркулес | 0,450 | Апельсин | 0,067 |
| кукуруза | 0,290 | Сыр адыгейский | 0,060 |
| лимон | 0,240 | Дыня | 0,047 |
Медикаментозное лечение [10,11]:
Современная патогенетическая терапия ГЛД основана на использовании медьэлиминирующих препаратов [10,11], главным образом Д-пеницилламина, триентина и солей цинка. Д-пеницилламин и триентин – хелатные комплексоны, образующие с медью прочные соединения, которые легко выводятся из организма с мочой.
Препаратом выбора при ГЛД является Д-пеницилламин. Лечение начинают с небольшой дозы с постепенным увеличением ее до терапевтической, под контролем выделения меди с мочой. Начальные дозы составляют 250-500 мг/сут с постепенным (каждые 4-7 дней) увеличением дозы на 250 мг до лечебной дозировки 1000-1500 мг/сут, которая дается в 2-4 приема. Д-пеницилламин назначается за 1 час или через 2 часа после приема пищи, т.к. еда снижает кишечную абсорбцию препарата. Когда клиническое состояние пациента стабилизируется, а экскреция меди с мочой снизится, дозу препарата уменьшают. На фоне приема Д-пеницилламина у больных со смешанной (неврологической) формой БВ может отмечаться ухудшение неврологической симптоматики, обусловленное индуцированной высокой мобилизации меди из печени и отложением ее в базальных ядрах головного мозга, что провоцирует или усиливает неврологическую симптоматику. Побочные явления на фоне терапии Д-пеницилламином (лихорадка, кожная сыпь, тромбоцитопения, лейкопения и пр.) наблюдаются у 25% пациентов. Возможно развитие подострой токсической реакции в виде протеинурии, угнетения костномозгового кровообращения или хронического токсического действия на кожу (преждевременное старение, дефекты в формировании рубцовой ткани, серпингинозный перфорирующий эластоз, вследствие токсического воздействия на коллагеновые волокна, возможно развитие слабости сосудистой стенки), иммунную систему с развитием аутоиммунных заболеваний (системная красная волчанка, артриты, повышение антинуклеарного фактора), а также снижение резистентности к инфекциям. Прием препарата прекращают, после исчезновения побочных эффектов возобновляют с минимальной дозы.
Д-пеницилламин является антагонистом пиридоксина, поэтому к терапии следует добавить пиридоксин в дозе 25-50 мг в сутки.
При выраженной и сохраняющейся непереносимости Д-пеницилламина назначаются препараты цинка.
Цинк (оксид, сульфат, ацетат) индуцирует синтез медьсвязывающих белков (металлотионинов) в эпителии тонкой кишки и гепатоцитах, что препятствует абсорбции меди в портальную циркуляцию. При лечении цинком медь выводится через кишечник. Рекомендуемая доза – 25 мг 3 раза в день, у детей и беременных в 2 раза меньше. Недостаточный ответ на лечение сопровождается повышенной экскрецией меди с мочой (более 125 мкг/сут). Показаниями к назначению препаратов цинка являются: стойкий резидуальный неврологический синдром, остающийся на фоне многолетней терапии Д- пеницилламином; обострение неврологической и (или) печеночной симптоматики в начальной стадии терапии Д-пеницилламина; доклиническая и доневрологическая стадии ГЛД. Лечение также проводится пожизненно.
| Лекарственная группа | Лекарственные средства | Способ применения | Уровень доказательности |
| Хелатор меди | Д-пеницилламин | 20мг/кг/сут в 2-4 приема, через 2ч после приема пищи. Последняя таблетка через 3ч после ужина. | В |
| Триентин * | Дети 12 лет и взрослые: 750—1250 мг/сут в 2—4 приема; максимальная доза 2 г/сут. | В | |
| Витамины | Пиридоксина гидрохлорид | 10мг по 1таблетке 2 раза в день после еды | С |
* применение препарата возможно после регистрации на территории РК.
| Препарат | Неврологическое ухудшение | Побочные эффекты | Комментарии |
| Д-пеницилламин | В начале лечения в 10-20% | -лихорадка, сыпь, протеинурия, волчаночноподобные реакции -апластическая анемия -лейкопения -тромбоцитопения -нефротический синдром -дегенеративные изменения кожи -серозный ретинит -гепатотоксичность | Уменьшение дозы при операциях для улучшения заживления раны и во время беременности. Уменьшение дозы на 25% при ремиссии |
| Цинк 1 | Возможно при начале лечения | -гастрит; биохимический панкреатит -аккумуляция цинка -возможные изменения в иммунной системе | Нет необходимости уменьшения дозы при операции или во время беременности |
| Триентин | В начале лечения в 10-15% | -гастрит — редко апластическая анемия -сидеробластная анемия | Уменьшение дозы при операциях для улучшения заживления раны и во время беременности. Уменьшение дозы на 25% при ремиссии |
1 нет клинических данных о совместном применении цинка с хелатирующим агентом
Таблица 9 — Перечень дополнительных лекарственных средств (менее 100% вероятности применения):
| Лекарственная группа | Лекарственные средства | Способ применения | Уровень доказательности |
| Индуктор металлотинеинов, блокирует всасывание меди в кишечнике | Цинк | По 25мг х 3 раза в день за 30мин до еды | С |
| Ингибитор протонной помпы | Пантопразол | старше 12 лет назначают по 40-80 мг в день | В |
| Эзомепразол | старше 12 лет 40-80 мг 1-2 раз в сутки | В | |
| Н2-гистаминоблокатор | Ранитидин | старше 14 лет – 150 мг 2 раза в сутки или 300 мг на ночь | С |
| Антихолинергические средства | Тригексифенидила гидрохлорид | 5-15 мг/сут, максимально до 40 мг/сут; в 2-3 приема | В |
| Атипичный нейролептик | Тиаприд | 3-6 мг/кг/сут в 2-3 приема | В |
| Производное бензодиазепина | Клоназепам | от 1 года до 5 лет — 1,5—3 мг в сутки; от 6 до 16 лет — 3—6 мг в сутки, в 2- 3 приёма. | В |
Хирургическое вмешательство:
1) Трансплантация печени
Показания:
· развитие фульминантной печеночной недостаточности;
· неэффективность терапии хелаторами меди в течение нескольких месяцев у пациентов с декомпенсированным циррозом печени;
· возникновение тяжелой прогрессирующей печеночной недостаточности при самостоятельном прекращении лечения, прогрессирующих и необратимых неврологических нарушениях.
2) Склеротерапия, облитерация, лигирование, прошивание варикозных вен пищевода
Показания:
· варикозное кровотечение.
Дальнейшее ведение:
· диспансерное наблюдение пожизненное (в первый год – ежемесячно с контролем лабораторных показателей; далее – 4 раза в год);
· на амбулаторном этапе после выписки из стационара: профилактика интеркуррентных заболеваний, диета с низким содержанием меди, обычный режим, дозированные физические упражнения;
· длительно сохраняющиеся соматические симптомы не являются противопоказанием к посещению школы; при выраженных неврологических и психических изменениях – обучение в специализированных школах или индивидуальное.
Индикаторы эффективности лечения:
· нормализация функциональных проб печени;
· уменьшение/исчезновение симптомов заболевания.
NB! Прогноз и исходы болезни Вильсона/
Болезнь Вильсона является прогрессирующим заболеванием и при отсутствии своевременной терапии больные умирают от осложнений цирроза печени и/или реже от прогрессирующей неврологической симптоматики. При хелирующей терапии и трансплантации печени длительная выживаемость пациентов с болезнью Вильсона стала нормой, хотя и не оценивалась проспективно.
Прогноз при болезни Вильсона связан со степенью декомпенсации печеночных функций, тяжестью неврологической симптоматики и приверженностью терапии. Нормализация печеночных функций происходит на 1-2 году терапии и не прогрессирует при полном выполнении всех рекомендаций. Консервативная терапия не эффективна при фульминантном течении заболевания. Был разработан прогностический индекс болезни Вильсона (Dhawan et al.), согласно которому оценка свыше 11 баллов связана с высокой вероятностью летального исхода при отсутствии срочной ортотопической трансплантации печени, таблица 10.
| Показатель | 1 балл | 2 балла | 3 балла | 4 балла |
| Билирубин (мкмоль/л) | 100-150 | 151-200 | 201-300 | >300 |
| АСТ (Ед/л) | 100-150 | 151-300 | 301-400 | >400 |
| МНО | 1.3-1.6 | 1.7-1.9 | 2.0-2.4 | >2.4 |
| Лейкоциты (10 9 ) | 6.8-8.3 | 8.4-10.3 | 10.4-15.3 | >15.3 |
| Альбумин г/л | 34-44 | 25-33 | 21-24 |
*-Суммарная оценка свыше 11 баллов связана с высокой вероятностью летального исхода без трансплантации печени.
Неврологическая симптоматика болезни Вильсона частично обратима при терапии хелаторами и проведении трансплантации печени, что связано с необратимыми поражениями подкорковых ядер головного мозга токсическими концентрациями меди.
Госпитализация
ПОКАЗАНИЯ ДЛЯ ГОСПИТАЛИЗАЦИИ С УКАЗАНИЕМ ТИПА ГОСПИТАЛИЗАЦИИ 1
Показания для плановой госпитализации [1-4]:
· для уточнения диагноза;
· подбор схемы терапии;
· декомпенсация, резистентность к лечению, побочные эффекты терапии.
· контроль эффективности терапии (оценка метаболизма меди, степень фиброзирования печеночной паренхимы и нарушения психо-неврологических функций, с определением показаний для своевременного выполнения ортотопической трансплантации печени.
Информация
Источники и литература
Информация
ОРГАНИЗАЦИОННЫЕ АСПЕКТЫ ПРОТОКОЛА
Список разработчиков протокола с указание квалификационных данных:
1) Шарипова Майра Набимуратовна – доктор медицинских наук, детский гастроэнтеролог клинико-диагностического отделения РГКП «Научный центр педиатрии и детской хирургии».
2) Карсыбекова Ляйля Мауленовна – доктор медицинских наук, профессор, педиатр клинико-диагностического отделения РГКП «Научный центр педиатрии и детской хирургии».
3) Мухамбетова Гульнар Амерзаевна – кандидат медицинских наук, профессор кафедры нервных болезней №1, РГП на ПХВ «Казахский национальный медицинский университет имени С.Д. Асфендиярова».
4) Калиева Шолпан Сабатаевна – кандидат медицинских наук, доцент, заведующая кафедрой клинической фармакологии и доказательной медицины РГП на ПХВ «Карагандинский государственный медицинский университет», клинический фармаколог.
Указание на отсутствие конфликта интересов: нет.
Рецензенты:
Кульниязова Гульшат Матаевна – доктор медицинских наук, профессор кафедры Общей врачебной практики №1 с курсом коммуникативных навыков РГП на ПХВ «Западно-Казахстанский государственный медицинский университет им. М. Оспанова».
Указание условий пересмотра протокола: пересмотр протокола через 5 лет после его опубликования или при наличии новых методов с уровнем доказательности.



